Introduction
|
|
Neuroembryology is a topic of the neurosciences that medical students have historically found one of the most challenging to comprehend [1][2][3][4][5][6][7][8][9][10][11][12]. Nevertheless, the subject is essential for future physicians to learn as it provides a framework into the understanding of human development [12]. Furthermore, it provides critical context to better appreciate congenital disorders of the brain and spinal cord.
A recent review of brainstem vascular syndromes demonstrated a simplified yet effective learning methodology for medical and health professional students [13]. In this brief review, we provide medical students an overview of the normal embryology of the nervous system as well as describe the common disorders that can occur when this process fails.
|
General Development of the Central Nervous System
|
|
The nervous system consists of three components. The brain and spinal cord comprise the central nervous system (CNS). The peripheral nervous system (PNS) consists of nerves outside of the CNS and includes the cranial nerves as well as spinal nerves. The autonomic nervous system is derived from both the CNS and PNS and consists of neurons innervating glands, cardiomyocytes and smooth musculature, among other structures.
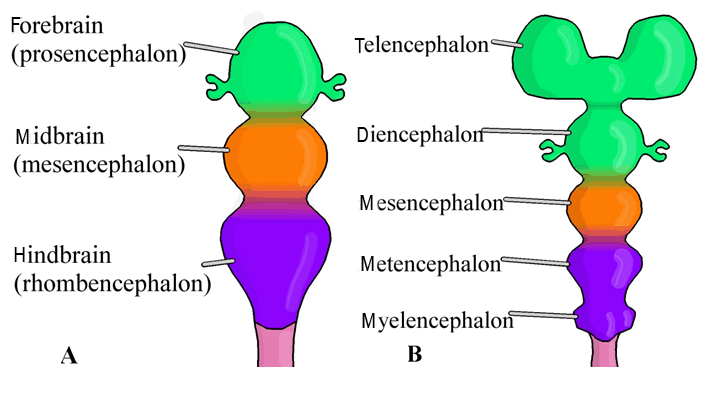
At approximately day-16 of embryological development, the notochord arises from a primary germ cell layer known as mesoderm [14][15][16]. The notochord induces the overlying ectoderm, another primary germ cell layer, to begin to form the neural plate [16]. Beginning in the third week, this neural plate represents the earliest form of the CNS, which will eventually separate into the brain and spinal cord (Figure 1A) [15][16]. The edges of the neural plate rise forming two parallel folds appropriately termed the neural folds ( (Figure 1B) [15] [16]. Continued elevation of the neural folds will result in fusion and formation of the neural tube (Figure 1C) [15][16]. Fusion of the neural folds occurs first in its middle and then proceeds to fuse in both the cranial and caudal directions giving rise to an opening at each end termed the cranial and caudal neuropores [14]. Complete closure of the cranial neuropore occurs on day-25 followed by closure of the caudal neuropore on day-28 [15]. During the fourth week, the cranial end of the neural tube exhibits three expansions known as the primary brain vesicles (Figure 2A) [16]. The primary vesicles include the prosencephalon, mesencephalon, and rhombencephalon that will eventually form the forebrain, midbrain and hindbrain, respectively [14][15][16]. Following the development of these primary vesicles, the secondary vesicles arise during the fifth week; that is, the three primary brain vesicles divide further into five secondary brain vesicles (Figure 2B) [16].
|
|
|
|
|
Neuroembryology of the Brain
|
|
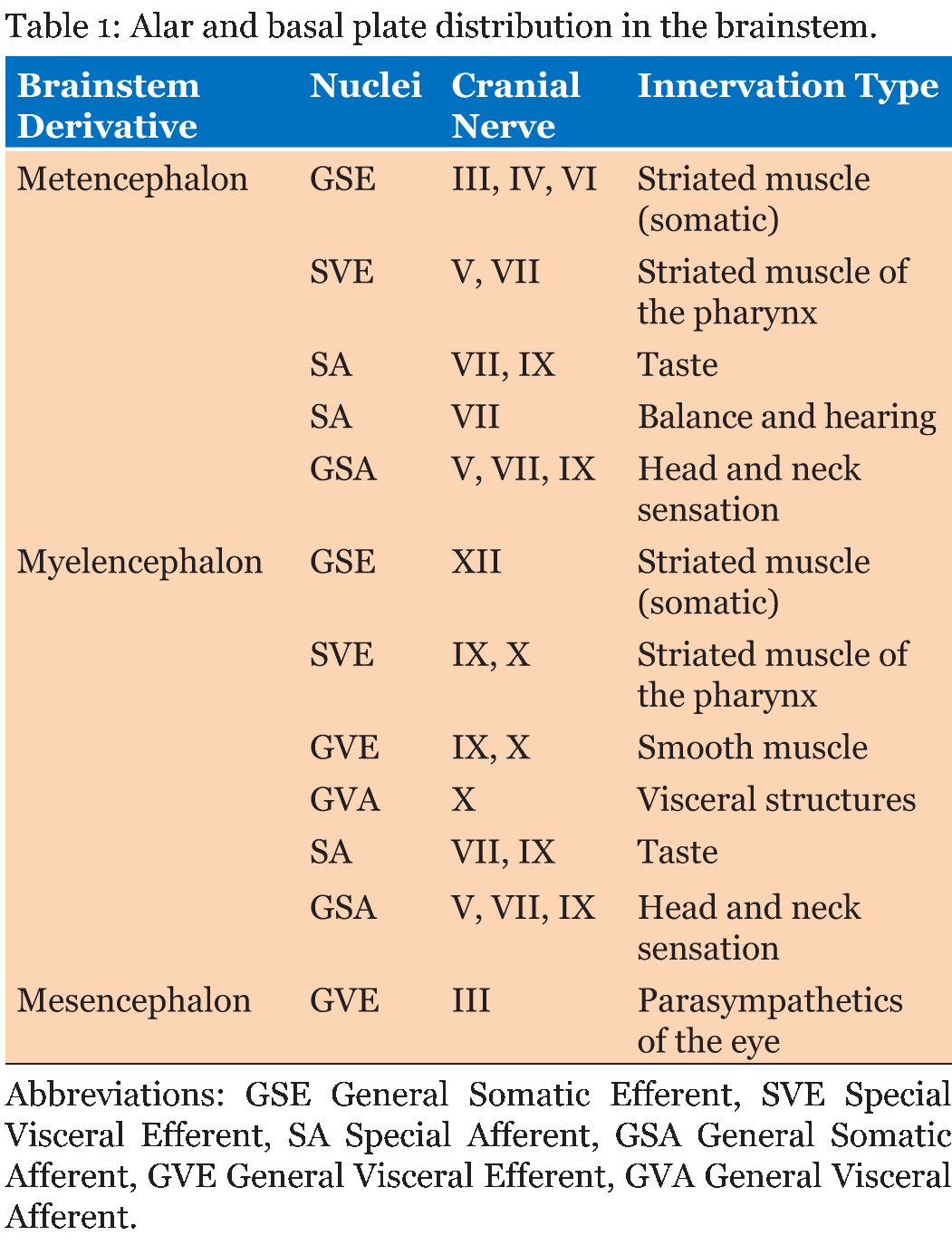
The rhombencephalon or hindbrain consists of the metencephalon and myelencephalon. The metencephalon forms the pons and cerebellum and contains the upper portion of the fourth ventricle. The myelencephalon forms the medulla oblongata and contains the lower portion of the fourth ventricle. The alar plates of the metencephalon and myelencephalon, which exist on the dorsal aspect of the neural tube, each consist of three groups of sensory nuclei. These groups of sensory nuclei include general visceral afferents, special afferents, and general somatic afferents (Table 1) [15]. The basal plates of the metencephalon and myelencephalon, which form on the ventral aspect of the neural tube, each consist of three groups of motor nuclei. These groups of motor nuclei include general visceral efferents, special efferents, and general somatic efferents [15].
Unlike the rhombencephalon and prosencephalon, the mesencephalon is a primary brain vesicle that does not divide further; rather, it forms the midbrain containing the aqueduct of Sylvius. The basal plates of the mesencephalon each consist of two groups of motor nuclei. These groups of sensory nuclei include general visceral efferents and general somatic efferents [15]. The prosencephalon or forebrain consists of the diencephalon and the telencephalon. The diencephalon forms the thalamus as well as the hypothalamus and contains the third ventricle and choroid plexus. Furthermore, this brain vesicle gives rise to the pineal body and neurohypophysis [15]. The telencephalon forms the cerebral hemispheres as well as the lamina terminalis and contains the lateral ventricles. The interventricular foramina of Monro of the diencephalon permits communication of cerebral spinal fluid between the lateral ventricles of the telencephalon.
|
|
|
Congenital Disorders of the Brain
|
|
Anencephaly
Anencephaly is the absence of a significant portion of the brain and skull. The failure of the neural tube to closes between day-23 and day-26 results in the disorder known as anencephaly [15] [17]. This neural tube disorder manifests phenotypically as an absence of the scalp, skull, and the majority of the brain. Despite its name literally translating to "no-head", usually only the telencephalon is absent [17]. Nevertheless, anencephaly is incompatible with life as most babies with this disorder expire within hours of birth. Clinical findings of this disorder include increased alpha-fetoprotein on maternal serum screening and polyhydramnios on 2nd and 3rd trimester ultrasound [18]. Although the etiology is uncertain, maternal folic acid deficiency has shown a strong correlation with the development of anencephaly.
Exencephaly
Exencephaly is a cephalic disorder where the brain is found outside of the skull. There is an absent cranial cavity and scalp with protruding brain tissue [19]. Additionally, there is an absence of the flat bones of the calvarium and bulging eyeballs. Exencephaly is considered a ciliopathy, meaning it is caused by aberrant cilia function around the 28th week of gestation [20]. Similar to anencephaly, exencephaly is also incompatible with life as the fetal brain cannot fully develop.
Cephalocele
A cephalocele is a protrusion of the meninges and/or neural tissue through an opening of the skull bones [19] [21]. It is thought that cephaloceles result from aberrant neural tube development [21]. Craniofacial abnormalities often present in patients with cephaloceles. Histologic examination permits classification of the cephalocele defined by the cranial contents that protrude through the skull. These include meningocele in which only the meninges is present outside the skull, pencehalomeningocele including both the meninges and cerebral tissue, encephalocystocele for a ventricular protrusion, and gliocele involving a glial-lined cyst with cerebrospinal fluid [15] [19][21]. All types of cephaloceles can lead to development of hydrocephalus, quadriparesis, seizures and severe mental retardation [19] [21].
Holoprosencephaly
Previously known as arhinencephaly, holoprosencephaly is a neural tube defect where the fetal prosencephalon fails to develop into two cerebral hemispheres [22]. Failure of the prosencephalon to develop restricts the division of the cerebral hemispheres resulting in craniofacial abnormalities such as cyclopia which is a midline defect classically typified in a solitary median eye [22]. Symptomatology of holoprosencephaly ranges from mild, presenting as a single central incisor, to cyclopia [23]. The exact etiology of this disorder remains uncertain; however, a mutation in the gene coding for Sonic Hedgehog protein has been shown to play a significant role [22]. Clinical signs include varying degrees of craniofacial abnormalities as well as seizures and mental retardation [22]
[23].
Schizencephaly
Schizencephaly is a disorder of cerebral cortical development [24]. This congenital disorder is characterized by clefts within the brain that are present from the pia mater to the ventricles of the cerebral hemispheres [24]. Clefts may be present unilaterally or bilaterally in the brain parenchyma [24]. Clefts are covered with dysplastic gray matter [24] [25]. The etiology of schizencephaly has been proven to be a cell migration defect. Patients usually present with microcephaly, hypotonia, mental retardation and hemiparesis or quadriparesis [24][25]. Prognosis is dependent on the size of the clefts within the brain parenchyma.
Lissencephaly
Lissencephaly meaning "smooth brain" is phenotypically characterized as a smooth surface of the brain's cerebral hemispheres [26]. There are two types of lissencephaly: type 1 known as classic lissencephaly and type 2 typified by a cobblestone pattern. Although both types of the disease have the same phenotypic manifestation, the etiologies differ. Type 1 is thought to be due to defective O-linked glycosylation of alpha-dystroglycan, which leads to a weak pial surface. The etiology of type 2 is less clearly defined. Clinical symptoms include hypotonia, muscle spasticity and most severely a failure to thrive. Prognosis for children varies depending on the severity of the disorder [26].
Hydranencephaly
Hydranencephaly is characterized as cerebral hemisphere necrosis and tremendous ventricular dilation [27]. The brain's cerebral hemispheres are absent to varying degrees and replaced by a membranous sac filled with cerebrospinal fluid [27]. Although the etiology is unclear, damage to the brain parenchyma is thought to be due to bilateral compromise of the internal carotid artery [27]. The classical hallmark of hydranencephaly is replacement of the brain parenchyma with cerebrospinal fluid resulting in transillumination of the neonatal skull [28]. Clinical manifestations include hypertonia, seizures and subsequent development of hydrocephalus [27][28]. Prognosis for hydranencephaly is generally poor with the majority of infants expiring before one year of age [27].
Dandy-Walker Syndrome
Dandy-Walker syndrome is a group of congenital brain malformations with three subclassifications all involving the cerebellum [29]. The key characteristics of this syndrome include fourth ventricle enlargement, cerebellar vermis aplasia and cyst formation [29]. Similar to exencephaly, Dandy-Walker syndrome is considered a ciliopathy [20]. This syndrome is often compatible with life; however, patients may exhibit mental retardation, slowed motor development and convulsions [29].
Joubert Syndrome
Joubert syndrome is a group of congenital anomalies of which the classic hallmark is a complex malformation of the midbrain and hindbrain known as the "molar tooth sign" [30]. Specifically, it indicates elongated superior cerebellar peduncles giving the midbrain an appearance of a molar tooth [30]. In addition to a malformed midbrain and hindbrain, Joubert syndrome classically involves underdevelopment or complete absence of the cerebellar vermis [30]. Similar to Dandy-Walker, Joubert syndrome is classified as a ciliopathy [20]. Typical clinical manifestations include cerebellar vermis hypoplasia, hypotonia and hyperpnea [20].
Pontine Tegmental Cap Dysplasia
First described in 2007, pontine tegmental cap dysplasia is a hindbrain malformation caused by defective neuronal migration or navigation of rhombencephalic neurons [31] [32]. This congenital malformation is characterized by ventral pons hypoplasia with a dorsal cap lying over the pontine tegmentum and bulging into the fourth ventricle [32][33]. Additional features of this disorder include cranial nerve deficits, cerebellar vermis hypoplasia and abnormalities of the cerebellar peduncles [33]. Clinically, patients can present with varying degrees of developmental delay, ataxia, horizontal gaze palsy and dysphagia [33].
Arnold-Chiari Malformations
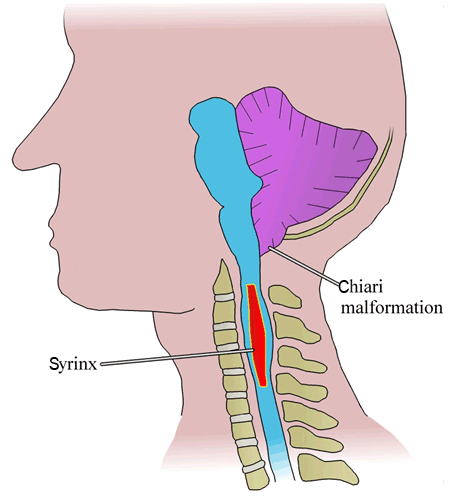
The final type of malformation that will be discussed is Arnold-Chiari malformations. These are classified into four types in order of increasing severity. The first three are characterized by downward displacement of the cerebellar tonsils through the foramen magnum of the skull (Figure 3) [34]. Type I usually presents solely with tonsillar herniation [34]. Chiari type I malformations may arise congenitally or can be acquired via trauma to the back of the head [35]. Interestingly, type I malformations are associated with connective tissue diseases such as Ehlers-Danlos syndrome [36]. Clinically, patients with type I can present with neck pain worsened by the Valsalva maneuver, ataxia, sleep apnea and predisposition to respiratory infections [37]. Type II is characterized by downward displacement of the cerebellar vermis and can be accompanied by myelomeningocele of the lumbosacral spine [37]. Type II will present clinically similar to type I, but with increased severity [37]. Furthermore, type II can present with paralysis below the level of the spinal defect [37][38]. Type III malformations are extremely rare, characterized by tonsillar herniation with occipital encephalocele [37]. In addition to the symptomatology described for type I and II malformations, type III can also present with seizures, spasticity and severe neurological deficits [37]. Contrary to the first three types of Chiari malformations, type IV does not present with hindbrain herniation; rather, this type presents with significant cerebellar and tentorial hypoplasia [37]. Type IV is the sole Chiari malformation that is always incompatible with life [34] [35] [36] [37] [38].
|
|
|
Neuroembryology of the Spinal Cord
|
|
After closure of the neural tube, neuroepithelial cells give rise to the primitive nerve cells known as neuroblasts [39]
[40][41]. Neuroblasts form a zone around the neuroepithelial cells, specifically known as the mantle layer of the spinal cord [15]. The mantle layer will eventually develop into the gray matter of the spinal cord. The outermost layer of the spinal cord called the marginal layer consists of nerves developing from the neuroblasts within the mantle layer [15]. Myelination of this layer will cause a white appearance of the tissue leading to what is formally known as the white matter [15]. Neuroblasts are continually added to the mantle layer resulting in a dorsal and ventral thickening of the neural tube [15] [16]. The ventral thickening is termed basal plates and consists of motor horn neurons [16] (Figure 4). The alar plates develop from the ventral thickening and consist of sensory neurons [16]. Additionally, neurons form an intermediate horn found between the ventral motor horn and dorsal sensory horn [16]. This intermediate horn contains sympathetic neurons and is present from the first thoracic spinal level through to the second and often third lumbar spinal level [15] [16].
|
|
|
Congenital Disorders of the Spinal Cord
|
|
Spina Bifida
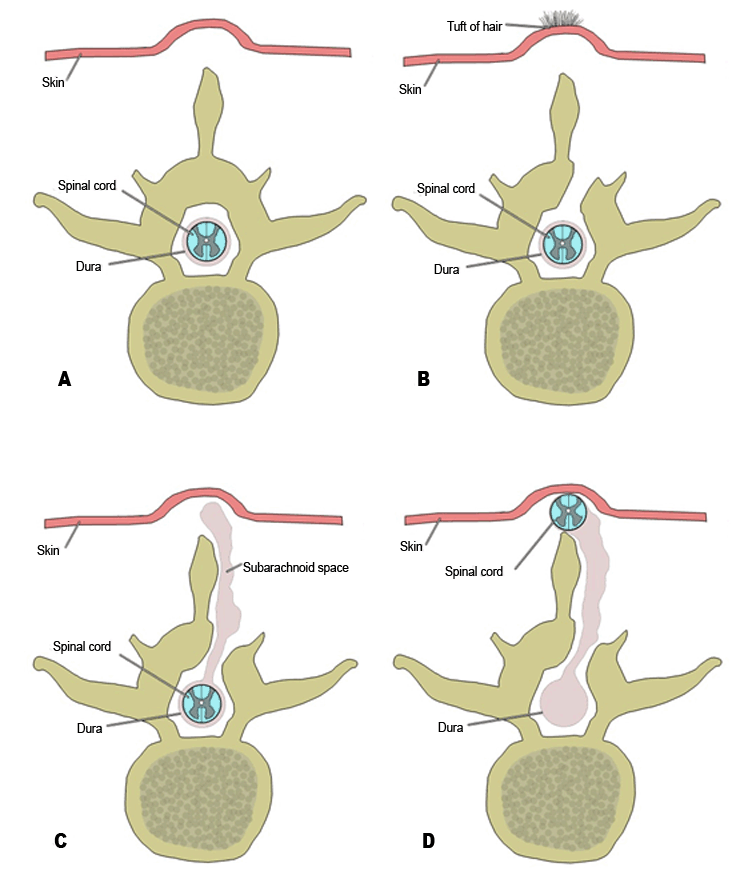
Abnormal closure of the neural folds results in a persistent connection between the amniotic cavity and the spinal canal [42]. This congenital malformation is commonly referred to as spina bifida. Neural tube defects can involve a variety of structures including the overlying skin, muscle, vertebrae, meninges and spinal cord itself. Spina bifida is further sub-classified dependent on the severity of the malformation (Figure 5A). Spina bifida occulta is a defect of the vertebral arches usually seen in the lower lumbar and sacral vertebral levels (Figure 5B). With spina bifida occulta, the skin and underlying dura are usually intact; however, it is associated with a patch of hair overlying the affected region [43]. A meningocele, the second subtype of spina bifida, is defined by herniation of the meninges, but not the neural tissue, through a vertebral arch defect (Figure 5C) [43]. Herniation of both the meninges and neural tissue through a bony defect is known as myelomeningocele (Figure 5D) [42]. The most severe form of spina bifida develops through failure of the neural folds to elevate during the third week of development. This results in the spinal cord external to the environment and is known as spina bifida with myeloschisis or rachischisis [44]. Fortunately, spina bifida is typically preventable with maternal intake of folic acid beginning at least one month prior to conception [45]. Elevations of alpha-fetoprotein in maternal serum screening and amniotic fluid allows for early screening of spina bifida with the exception of occulta. Moreover, acetylcholinesterase can be found elevated in the amniotic fluid making it a helpful secondary biomarker [46].
Syringomyelia
Syringomyelia is characterized by a fluid-filled cavity beginning in the central canal or spinal cord tissue (Figure 3) [47]. This condition can arise congenitally or it can be acquired later in life. An acquired syringomyelia occurs as a complication of trauma, tumor, hemorrhage or inflammation of the meninges [47]. A cyst, or syrinx develops in an area of spinal cord damage and further expands causing progressive neurologic symptomatology including weakness, pain, and sensory impairment [48]. Congenital syringomyelia is commonly associated with type I Arnold Chiari malformations as previously described [47].
|
|
|
Neuroembryology of the Cranial Nerves
|
|
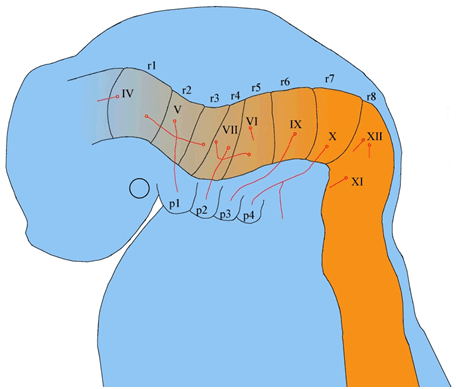
By the end of the fourth week, cranial nuclei have developed from the brain stem with the exception of the olfactory and optic nerves, which develop from the forebrain [15]. Apart from the oculomotor nuclei, the remaining 9 cranial nerves arise from the hindbrain [15]. Here, eight discrete segments develop, known as the rhombomeres which form the motor nuclei of cranial nerves IV, V, VI, VII, IX, X, XI, and XII (Figure 6) [39]. Despite not all cranial nerves consisting of motor and sensory nerve fibers, the configuration of cranial nerves is analogous to spinal nerves [15]. Motor neurons of cranial nerve nuclei exist in the brainstem, whereas the sensory ganglia exist outside of the brain [16]. The sensory ganglia of cranial nerves are derived from ectodermal placodes and neural crest cells [15][40]. A neurogenic placode is thickened epithelium of ectodermal layer, which gives rise to the sensory aspect of the nervous system [15] [40].
|
|
|
Congenital Disorders of the Cranial Nerves
|
|
Congenital Cranial Dysinnervation Disorders
Congenital cranial dysinnervation disorders include several similar conditions such as Duane retraction syndrome, hereditary congenital facial paresis, Mobius syndrome, congenital blepharoptosis and congenital fibrosis of the external ocular muscles [49]. The characteristic etiology in these congenital disorders is loss of innervation to motor muscles. Furthermore, these disorders are non-progressive in nature. Duane syndrome presents with limited horizontal eye movement as well as eye retraction when adduction is attempted [50]. Hereditary congenital facial paresis is a syndrome of facial nerve palsy resulting in ptosis and family asymmetry [51]. Mobius syndrome is characterized as facial weakness and restricted ocular abduction [49]. Congenital blepharoptosis presents with either unilateral or bilateral ptosis [52]. Congenital fibrosis of the external ocular muscles is a group of eye movement disorders resulting from dysfunction of the oculomotor and trochlear nerves [53].
|
|
Congenital Disorders of the Autonomic Nervous System
|
|
Hirschsprung Disease
Hirschsprung disease is a type of megacolon characterized by the absence of all or part of the enteric nervous system within the distal gut [54] Known as congenital aganglionic megacolon. In normal development, neural crest cells migrate to the colon forming the myenteric plexus and the submucosal plexus. The failure of migration of these neural crest cells results in an aganglionic segment of the colon [54]. Improper peristalsis of colonic mass movements results in obstruction of the colon [54]. Hirschsprung disease is typically diagnosed soon after birth with failure to pass meconium within the first 48 hours after delivery [54]. Definitive diagnosis is made by suction biopsy of the aganglionic segment [55].
|
Conclusion
|
|
Herein, we have provided a brief overview of the neuroembryology and the subsequent congenital disorders of the nervous system. Neuroembryology is an important yet menacing subject for early medical students and practicing clinicians alike. Like all congenital pathophysiology, one must understand the 'normal' development before diagnosing the 'abnormal'. Therefore, a basic working knowledge of neuroembryology is essential for future physicians to acquire as it allows one to better understand the congenital disorders that will be encountered in the hospital setting.
|
References
|
-
Lim EC, Seet RC. Demystifying neurology: preventing 'neurophobia' among medical students. Nat Clin Pract Neurol 2008 Aug;4(8):462–3.
[CrossRef]
[Pubmed]

-
McGee J, Maghzi AH, Minagar A. Neurophobia: A global and under-recognized phenomenon. Clin Neurol Neurosurg 2014 Jul;122:iii-iv.
[CrossRef]
[Pubmed]
-
Ramos RL, Smith PT. A core neuroanatomy syllabus for diverse student populations. Clin Anat 2016 Mar;29(2):131.
[CrossRef]
[Pubmed]
-
Zinchuk AV, Flanagan EP, Tubridy NJ, Miller WA, McCullough LD. Attitudes of US medical trainees towards neurology education: "Neurophobia" - a global issue. BMC Med Educ 2010 Jun 23;10:49.
[CrossRef]
[Pubmed]
-
Pakpoor J, Handel AE, Disanto G, et al. National survey of UK medical students on the perception of neurology. BMC Med Educ 2014 Oct 21;14:225.
[CrossRef]
[Pubmed]
-
Flanagan E, Walsh C, Tubridy N. 'Neurophobia'–attitudes of medical students and doctors in Ireland to neurological teaching. Eur J Neurol 2007 Oct;14(10):1109–12.
[CrossRef]
[Pubmed]
-
Lukas RV, Cooper B, Morgan I, Brorson JR, Dong H, Sherer R. Attitudes toward neurosciences in medical students in Wuhan, China: A survey study. World Neurosurg 2014 Sep-Oct;82(3-4):266–9.
[CrossRef]
[Pubmed]
-
Matthias AT, Nagasingha P, Ranasinghe P, Gunatilake SB. Neurophobia among medical students and non-specialist doctors in Sri Lanka. BMC Med Educ 2013 Dec 9;13:164.
[CrossRef]
[Pubmed]
-
Youssef FF. Neurophobia and its implications: Evidence from a Caribbean medical school. BMC Med Educ 2009 Jul 1;9:39.
[CrossRef]
[Pubmed]
-
Ramos RL, Cuoco JA, Guercio E, Levitan T. Quantitative Description of Medical Student Interest in Neurology and Psychiatry. J Am Osteopath Assoc 2016 Jul 1;116(7):462–71.
[CrossRef]
[Pubmed]
-
Cuoco JA. Medical student neurophobia: A review of the current pandemic and proposed educational solutions. European Journal of Educational Sciences 2016 Sept;3(3):41–6.
-
Carlson BM. Embryology in the medical curriculum. Anat Rec 2002 Apr 15;269(2):89–98.
[CrossRef]
[Pubmed]
-
Cuoco JA, Hitscherich K, Hoehmann CL. Brainstem vascular syndromes: A practical guide for medical students. Edorium J Neurol 2016 Sept;3(1):4–16.
-
Greene ND, Copp AJ. Development of the vertebrate central nervous system: formation of the neural tube. Prenat Diagn 2009 Apr;29(4):303–11.
[CrossRef]
[Pubmed]
-
Sadler TW. Langman's medical embryology. 12ed. Philadelphia: Lippincott Williams & Wilkins; 2012.
-
Haines DE. Fundamental neuroscience for basic and clinical applications. 4ed. Philadelphia: Elsevier; 2013.
-
Barron S. Anencephaly: An Ongoing Investigation in Washington State. Am J Nurs 2016 Mar;116(3):60–6.
[CrossRef]
[Pubmed]
-
Verity C, Firth H, ffrench-Constant C. Congenital abnormalities of the central nervous system. J Neurol Neurosurg Psychiatry 2003 Mar;74 Suppl 1:i3–8.
[CrossRef]
[Pubmed]
-
Nuñez S, Mantilla MT, Bermúdez S. Midline congenital malformations of the brain and skull. Neuroimaging Clin N Am 2011 Aug;21(3):429–82, vii.
[CrossRef]
[Pubmed]
-
Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 2006;7:125–48.
[CrossRef]
[Pubmed]
-
Raja RA, Qureshi AA, Memon AR, Ali H, Dev V. Pattern of encephaloceles: a case series. J Ayub Med Coll Abbottabad 2008 Jan-Mar;20(1):125–8.
[Pubmed]
-
Kauvar EF, Muenke M. Holoprosencephaly: recommendations for diagnosis and management. Curr Opin Pediatr 2010 Dec;22(6):687–95.
[CrossRef]
[Pubmed]
-
Raybaud C, Widjaja E. Development and dysgenesis of the cerebral cortex: malformations of cortical development. Neuroimaging Clin N Am 2011 Aug;21(3):483–543, vii.
[CrossRef]
[Pubmed]
-
Halabuda A, Klasa L, Kwiatkowski S, Wyrobek L, Milczarek O, Gergont A. Schizencephaly-diagnostics and clinical dilemmas. Childs Nerv Syst 2015 Apr;31(4):551–6.
[CrossRef]
[Pubmed]
-
Dies KA, Bodell A, Hisama FM, et al. Schizencephaly: association with young maternal age, alcohol use, and lack of prenatal care. J Child Neurol 2013 Feb;28(2):198–203.
[CrossRef]
[Pubmed]
-
Mochida GH. Genetics and biology of microcephaly and lissencephaly. Semin Pediatr Neurol 2009 Sep;16(3):120–6.
[CrossRef]
[Pubmed]
-
Cecchetto G, Milanese L, Giordano R, Viero A, Suma V, Manara R. Looking at the missing brain: hydranencephaly case series and literature review. Pediatr Neurol 2013 Feb;48(2):152–8.
[CrossRef]
[Pubmed]
-
Barozzino T, Sgro M. Transillumination of the neonatal skull: seeing the light. CMAJ 2002 Nov 26;167(11):1271–2.
[Pubmed]
-
McClelland S 3rd, Ukwuoma OI, Lunos S, Okuyemi KS. The natural history of Dandy-Walker syndrome in the United States: A population-based analysis. J Neurosci Rural Pract 2015 Jan;6(1):23–6.
[CrossRef]
[Pubmed]
-
Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010 Jul 8;5:20.
[CrossRef]
[Pubmed]
-
Barth PG, Majoie CB, Caan MW, et al. Pontine tegmental cap dysplasia: A novel brain malformation with a defect in axonal guidance. Brain 2007 Sep;130(Pt 9):2258–66.
[CrossRef]
[Pubmed]
-
Szczaluba K, Szymanska K, Bekiesinska-Figatowska M, et al. Pontine tegmental cap dysplasia: A hindbrain malformation caused by defective neuronal migration. Neurology 2010 Jun 1;74(22):1835.
[CrossRef]
[Pubmed]
-
Harding B, Vossough A, Goldberg E, Santi M. Pontine tegmental cap dysplasia: neuropathological confirmation of a rare clinical/radiological syndrome. Neuropathol Appl Neurobiol 2016 Apr;42(3):301–6.
[CrossRef]
[Pubmed]
-
Arnett B. Arnold-Chiari malformation. Arch Neurol 2003 Jun;60(6):898–900.
[CrossRef]
[Pubmed]
-
Mora JR, Rison RA, Beydoun SR. Chiari malformation type I with cervicothoracic syringomyelia masquerading as bibrachial amyotrophy: A case report. J Med Case Rep 2015 Jan 27;9:11.
[CrossRef]
[Pubmed]
-
Castori M, Voermans NC. Neurological manifestations of Ehlers-Danlos syndrome(s): A review. Iran J Neurol 2014 Oct 6;13(4):190–208.
[Pubmed]
-
Vannemreddy P, Nourbakhsh A, Willis B, Guthikonda B. Congenital Chiari malformations. Neurol India 2010 Jan-Feb;58(1):6–14.
[CrossRef]
[Pubmed]
-
Ganesh D, Sagayaraj BM, Barua RK, Sharma N, Ranga U. Arnold Chiari malformation with spina bifida: a lost opportunity of folic Acid supplementation. J Clin Diagn Res 2014 Dec;8(12):OD01–3.
[CrossRef]
[Pubmed]
-
Danowitz M, Zheng H, Guigova A, Solounias N. A combined approach of teaching head development using embryology and comparative anatomy. Edorium J Anat Embryo 2016;3:17–27.
-
Graham A, Shimeld SM. The origin and evolution of the ectodermal placodes. J Anat 2013 Jan;222(1):32–40.
[CrossRef]
[Pubmed]
-
Akizu N, García MA, Estarás C, et al. EZH2 regulates neuroepithelium structure and neuroblast proliferation by repressing p21. Open Biol 2016 Apr;6(4):150227.
[CrossRef]
[Pubmed]
-
Sandler AD. Children with spina bifida: key clinical issues. Pediatr Clin North Am 2010 Aug;57(4):879–92.
[CrossRef]
[Pubmed]
-
Liptak GS, Garver K, Dosa NP. Spina bifida grown up. J Dev Behav Pediatr 2013 Apr;34(3):206–15.
[CrossRef]
[Pubmed]
-
Jeelani Y, McComb JG. Congenital hydrocephalus associated with myeloschisis. Childs Nerv Syst 2011 Oct;27(10):1585–8.
[CrossRef]
[Pubmed]
-
Ahrens K, Yazdy MM, Mitchell AA, Werler MM. Folic acid intake and spina bifida in the era of dietary folic acid fortification. Epidemiology 2011 Sep;22(5):731–7.
[CrossRef]
[Pubmed]
-
Loft AG, Høgdall E, Larsen SO, Nørgaard-Pedersen B. A comparison of amniotic fluid alpha-fetoprotein and acetylcholinesterase in the prenatal diagnosis of open neural tube defects and anterior abdominal wall defects. Prenat Diagn 1993 Feb;13(2):93–109.
[Pubmed]
-
Greitz D. Unraveling the riddle of syringomyelia. Neurosurg Rev 2006 Oct;29(4):251–63; discussion 264.
[CrossRef]
[Pubmed]
-
Nakamura M, Ishii K, Watanabe K, et al. Clinical significance and prognosis of idiopathic syringomyelia. J Spinal Disord Tech 2009 Jul;22(5):372–5.
[CrossRef]
[Pubmed]
-
Gutowski NJ, Chilton JK. The congenital cranial dysinnervation disorders. Arch Dis Child 2015 Jul;100(7):678–81.
[CrossRef]
[Pubmed]
-
Kalevar A, Ong Tone S, Flanders M. Duane syndrome: Clinical features and surgical management. Can J Ophthalmol 2015 Aug;50(4):310–3.
[CrossRef]
[Pubmed]
-
Alrashdi IS, Rich P, Patton MA. A family with hereditary congenital facial paresis and a brief review of the literature. Clin Dysmorphol 2010 Oct;19(4):198–201.
[CrossRef]
[Pubmed]
-
SooHoo JR, Davies BW, Allard FD, Durairaj VD. Congenital ptosis. Surv Ophthalmol 2014 Sep-Oct;59(5):483–92.
[CrossRef]
[Pubmed]
-
Cooymans P, Al-Zuhaibi S, Al-Senawi R, Ganesh A. Congenital fibrosis of the extraocular muscles. Oman J Ophthalmol 2010 May;3(2):70–4.
[CrossRef]
[Pubmed]
-
Kenny SE, Tam PK, Garcia-Barcelo M. Hirschsprung's disease. Semin Pediatr Surg 2010 Aug;19(3):194–200.
[CrossRef]
[Pubmed]
-
Rahman Z, Hannan J, Islam S. Hirschsprung's disease: Role of rectal suction biopsy - data on 216 specimens. J Indian Assoc Pediatr Surg 2010 Apr;15(2):56–8.
[CrossRef]
[Pubmed]
|