 |
|
Review Article
| ||||||
| Neuroembryology and congenital disorders of the nervous system: A primer for medical students | ||||||
| Joshua A. Cuoco1, Christopher L. Hoehmann2, Dane M. Edwards3 | ||||||
|
1MS,Third year medical student, Department of Biomedical Sciences, New York Institute of Technology College of Osteopathic Medicine, Old Westbury, New York 11568 USA.
2BS,Third year medical student, Department of Anatomy, New York Institute of Technology College of Osteopathic Medicine, Old Westbury, New York 11568 USA. 3BS, Third year medical student, Faculty of Medicine, University of British Columbia, Vancouver, Canada. | ||||||
| ||||||
|
[HTML Abstract]
[PDF Full Text]
[Print This Article]
[Similar article in Pumed] [Similar article in Google Scholar] |


| How to cite this article |
| Cuoco JA, Hoehmann CL, Edwards DM. Neuroembryology and congenital disorders of the nervous system: A primer for medical students. Edorium J Neurol 2016;3:17–25. |
|
Abstract
|
|
Embryonic development of the nervous system is a challenging topic within the medical school curriculum. Nonetheless, it is essential for future physicians to have a basic knowledge of this subject as it provides context for the congenital disorders affecting the nervous system. Supplemented with numerous illustrations, this article provides medical students a basic context of homeostatic neuroembryology of the brain, spinal cord and cranial nerves. Furthermore, we provide a brief overview of the common congenital disorders that can occur as a result of disruptions in these normal developmental processes.
| |
|
Keywords:
Medical Education, Neurodevelopment, Neuroembryology, Neurophobia
| |
|
Introduction
|
|
Neuroembryology is a topic of the neurosciences that medical students have historically found one of the most challenging to comprehend [1][2][3][4][5][6][7][8][9][10][11][12]. Nevertheless, the subject is essential for future physicians to learn as it provides a framework into the understanding of human development [12]. Furthermore, it provides critical context to better appreciate congenital disorders of the brain and spinal cord. A recent review of brainstem vascular syndromes demonstrated a simplified yet effective learning methodology for medical and health professional students [13]. In this brief review, we provide medical students an overview of the normal embryology of the nervous system as well as describe the common disorders that can occur when this process fails. |

|
Neuroembryology of the Brain
| ||||||
|
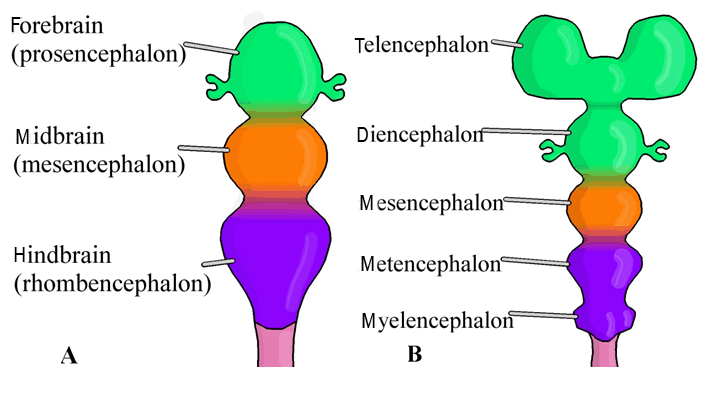
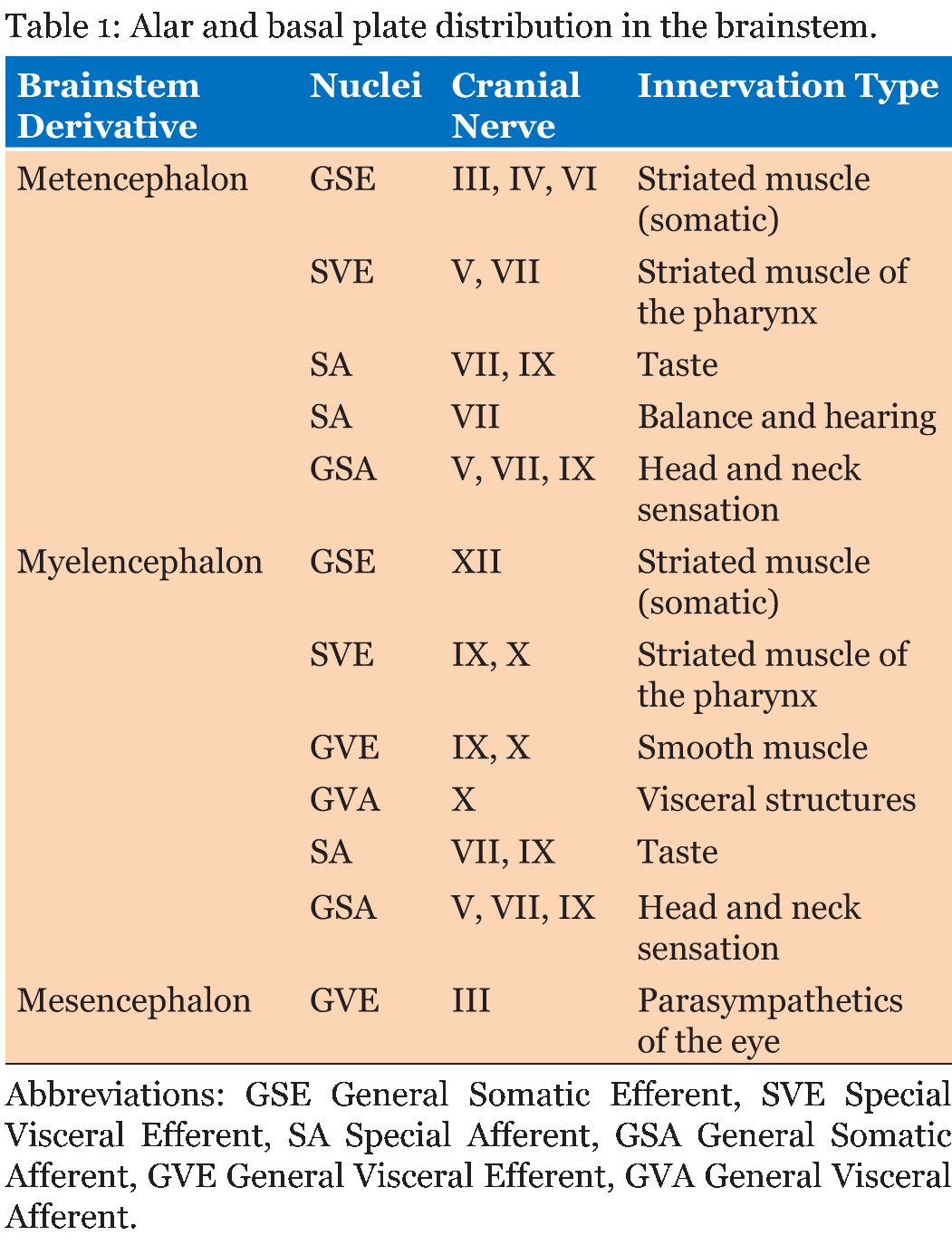
The rhombencephalon or hindbrain consists of the metencephalon and myelencephalon. The metencephalon forms the pons and cerebellum and contains the upper portion of the fourth ventricle. The myelencephalon forms the medulla oblongata and contains the lower portion of the fourth ventricle. The alar plates of the metencephalon and myelencephalon, which exist on the dorsal aspect of the neural tube, each consist of three groups of sensory nuclei. These groups of sensory nuclei include general visceral afferents, special afferents, and general somatic afferents (Table 1) [15]. The basal plates of the metencephalon and myelencephalon, which form on the ventral aspect of the neural tube, each consist of three groups of motor nuclei. These groups of motor nuclei include general visceral efferents, special efferents, and general somatic efferents [15]. Unlike the rhombencephalon and prosencephalon, the mesencephalon is a primary brain vesicle that does not divide further; rather, it forms the midbrain containing the aqueduct of Sylvius. The basal plates of the mesencephalon each consist of two groups of motor nuclei. These groups of sensory nuclei include general visceral efferents and general somatic efferents [15]. The prosencephalon or forebrain consists of the diencephalon and the telencephalon. The diencephalon forms the thalamus as well as the hypothalamus and contains the third ventricle and choroid plexus. Furthermore, this brain vesicle gives rise to the pineal body and neurohypophysis [15]. The telencephalon forms the cerebral hemispheres as well as the lamina terminalis and contains the lateral ventricles. The interventricular foramina of Monro of the diencephalon permits communication of cerebral spinal fluid between the lateral ventricles of the telencephalon. | ||||||
| ||||||
|
| ||||||
|
Congenital Disorders of the Brain
| ||||||
|
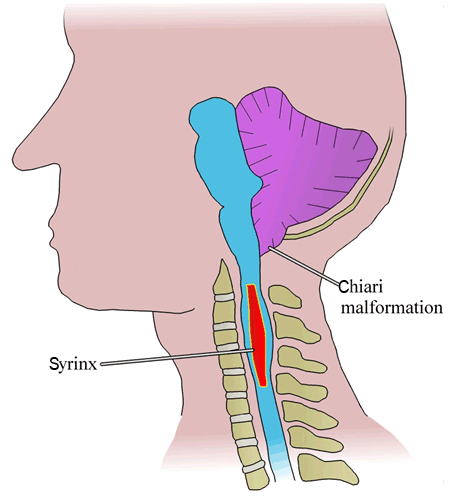
Anencephaly Exencephaly Cephalocele Holoprosencephaly Schizencephaly Lissencephaly Hydranencephaly Dandy-Walker Syndrome Joubert Syndrome Pontine Tegmental Cap Dysplasia Arnold-Chiari Malformations | ||||||
| ||||||
|
| ||||||
|
Neuroembryology of the Spinal Cord
| ||||||
|
After closure of the neural tube, neuroepithelial cells give rise to the primitive nerve cells known as neuroblasts [39] [40][41]. Neuroblasts form a zone around the neuroepithelial cells, specifically known as the mantle layer of the spinal cord [15]. The mantle layer will eventually develop into the gray matter of the spinal cord. The outermost layer of the spinal cord called the marginal layer consists of nerves developing from the neuroblasts within the mantle layer [15]. Myelination of this layer will cause a white appearance of the tissue leading to what is formally known as the white matter [15]. Neuroblasts are continually added to the mantle layer resulting in a dorsal and ventral thickening of the neural tube [15] [16]. The ventral thickening is termed basal plates and consists of motor horn neurons [16] (Figure 4). The alar plates develop from the ventral thickening and consist of sensory neurons [16]. Additionally, neurons form an intermediate horn found between the ventral motor horn and dorsal sensory horn [16]. This intermediate horn contains sympathetic neurons and is present from the first thoracic spinal level through to the second and often third lumbar spinal level [15] [16]. | ||||||
| ||||||
|
| ||||||

|
Congenital Disorders of the Spinal Cord
| ||||||
|
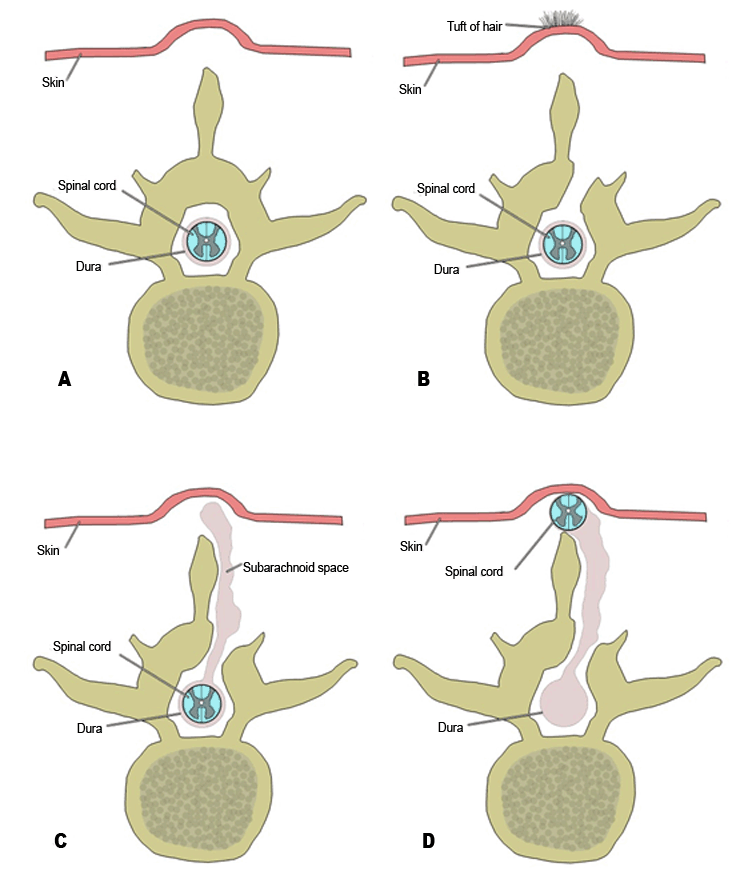
Spina Bifida Syringomyelia | ||||||
| ||||||
|
| ||||||
|
Neuroembryology of the Cranial Nerves
| ||||||
|
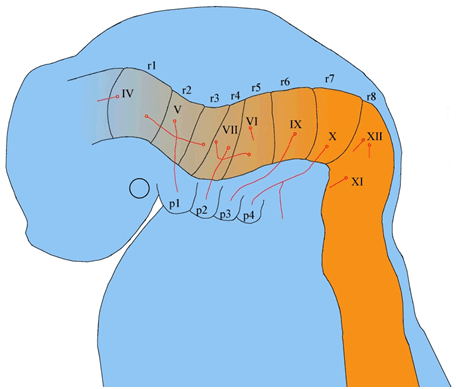
By the end of the fourth week, cranial nuclei have developed from the brain stem with the exception of the olfactory and optic nerves, which develop from the forebrain [15]. Apart from the oculomotor nuclei, the remaining 9 cranial nerves arise from the hindbrain [15]. Here, eight discrete segments develop, known as the rhombomeres which form the motor nuclei of cranial nerves IV, V, VI, VII, IX, X, XI, and XII (Figure 6) [39]. Despite not all cranial nerves consisting of motor and sensory nerve fibers, the configuration of cranial nerves is analogous to spinal nerves [15]. Motor neurons of cranial nerve nuclei exist in the brainstem, whereas the sensory ganglia exist outside of the brain [16]. The sensory ganglia of cranial nerves are derived from ectodermal placodes and neural crest cells [15][40]. A neurogenic placode is thickened epithelium of ectodermal layer, which gives rise to the sensory aspect of the nervous system [15] [40]. | ||||||
| ||||||
|
| ||||||
|
Congenital Disorders of the Cranial Nerves
|
|
Congenital Cranial Dysinnervation Disorders |
|
|
|
Congenital Disorders of the Autonomic Nervous System
|
|
Hirschsprung Disease |
|
Conclusion |
|
Herein, we have provided a brief overview of the neuroembryology and the subsequent congenital disorders of the nervous system. Neuroembryology is an important yet menacing subject for early medical students and practicing clinicians alike. Like all congenital pathophysiology, one must understand the 'normal' development before diagnosing the 'abnormal'. Therefore, a basic working knowledge of neuroembryology is essential for future physicians to acquire as it allows one to better understand the congenital disorders that will be encountered in the hospital setting. |
|
References
|
|

|
[HTML Abstract]
[PDF Full Text]
|
|
Author Contributions
Joshua A. Cuoco – Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Christopher L. Hoehmann – Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published Dane M. Edwards – Conception and design, Acquisition of data, Analysis and interpretation of data, Drafting the article, Critical revision of the article, Final approval of the version to be published |
|
Guarantor of submission
The corresponding author is the guarantor of submission. |
|
Source of support
None |
|
Conflict of interest
Authors declare no conflict of interest. |
|
Copyright
© 2016 Joshua A. Cuoco et al. This article is distributed under the terms of Creative Commons Attribution License which permits unrestricted use, distribution and reproduction in any medium provided the original author(s) and original publisher are properly credited. Please see the copyright policy on the journal website for more information. |
|
|